[本站讯]近日,齐鲁医院络病理论创新转化全国重点实验室张澄教授、张猛教授和张文程教授研究团队在心血管疾病基础研究领域中,发现了血管内皮功能异常、动脉粥样硬化(AS)和急性心肌梗死(AMI)的系列发病新机制和干预新靶点,研究成果发表于美国心脏学会(AHA)顶刊Circulation(中科院1区Top期刊,IF:38.6,2篇)和欧洲心脏协会(ESC)顶刊European Heart Journal(中科院1区Top期刊,IF:35.6,2篇)。
动脉粥样硬化是冠心病、缺血性脑卒中等心脑血管疾病的共同病理学基础,其核心病理特征为动脉内膜下脂质沉积、免疫细胞浸润和慢性炎症反应。巨噬细胞作为炎症反应的关键细胞,通过摄取氧化低密度脂蛋白(ox-LDL)形成泡沫细胞,释放促炎因子,推动斑块形成、进展与不稳定,是心血管疾病发生和发展的重要驱动力。然而,在持续的脂负荷和炎症微环境中,巨噬细胞是否存在内源性的“保护性机制”以及这一机制在疾病进展中为何失效尚不明确。
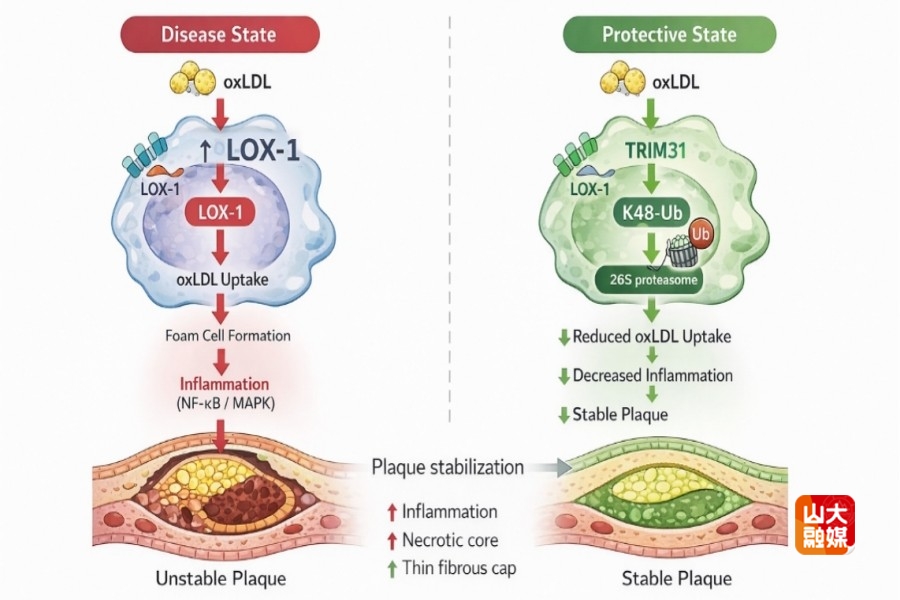
针对上述问题,齐鲁医院全国重点实验室的张澄教授和张猛教授团队通过转录组学筛选并在单核细胞/巨噬细胞中验证发现,E3泛素连接酶TRIM31在脂质刺激下呈显著的时间和剂量依赖性升高,提示TRIM31可能参与巨噬细胞介导的脂质应答与炎症调控。该团队构建了巨噬细胞特异性TRIM31缺失小鼠,发现巨噬细胞TRIM31缺失显著增加了主动脉斑块面积,同时伴随坏死核心扩大、胶原含量下降、多种炎症因子表达显著上调、纤维帽变薄等典型不稳定AS斑块的特征。在体外研究中,该团队进一步证实,在ox-LDL刺激条件下,TRIM31缺失的巨噬细胞表现出更强的脂质内吞及泡沫化,同时促炎因子的表达与分泌上调。相反,过表达TRIM31可显著抑制ox-LDL诱导的巨噬细胞脂质摄取与泡沫细胞形成,并降低促炎因子水平,提示TRIM31可在细胞水平限制脂质负荷和炎症反应。在机制研究中,该团队结合免疫共沉淀-质谱(IP-MS)、差异蛋白组学及体内外泛素化实验,鉴定出氧化低密度脂蛋白受体LOX-1是TRIM31的重要靶蛋白。进一步研究发现,TRIM31可直接与LOX-1相互作用,介导LOX-1第12位赖氨酸残基的K48-连接型泛素化修饰,从而促进LOX-1经蛋白酶体途径的降解。该过程不影响LOX-1的转录水平,而是通过调控其蛋白稳定性,限制巨噬细胞对ox-LDL的摄取。该项研究揭示了TRIM31作为AS内源性保护因子的新机制,提出TRIM31–LOX-1轴可能成为动脉粥样硬化性心血管疾病(ASCVD)中巨噬细胞靶向治疗的重要干预节点。该研究发表于Circulation,山东大学齐鲁医院心血管内科副教授张杰、博士后俞力雯是该文的共同第一作者,张澄教授、张猛教授为该文的共同通讯作者。山东大学齐鲁医院为第一和通讯作者单位。
随着AS免疫调控机制的逐步阐明,AS已被视为一种慢性炎症性疾病。尽管AS的降脂治疗已取得显著成效,但ASCVD的残余炎症风险仍是临床治疗的重大挑战。巨噬细胞介导的炎症反应是驱动AS斑块进展的核心环节,其中NLRP3炎症小体的过度激活扮演了关键角色。然而,巨噬细胞是否存在内源性的“刹车”机制以精确调控NLRP3炎症小体的活性,以及如何利用这一机制进行安全有效的干预,仍是亟待解决的科学难题。
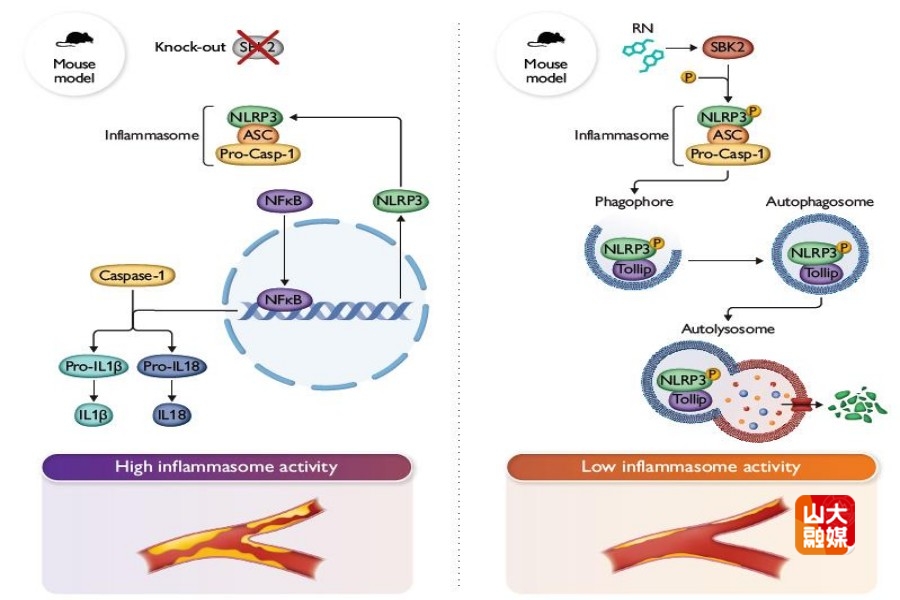
针对上述问题,山东大学齐鲁医院全国重点实验室的张澄教授和张猛教授团队通过分析高脂饮食喂养不同时间点的小鼠AS斑块的单细胞测序数据,发现在晚期斑块的巨噬细胞中,一种此前研究甚少的激酶SBK2表达显著上调。继之,在人和小鼠AS病变的验证研究中发现,SBK2在巨噬细胞中特异性高表达,且表达水平与疾病严重程度和血清促炎因子水平呈正相关。为了明确SBK2在AS发生和发展中的作用,该团队构建了全身性和巨噬细胞特异性SBK2敲除小鼠模型。在高脂饮食诱导下,SBK2缺失显著加剧了主动脉斑块负荷,表现为斑块内脂质沉积、巨噬细胞浸润、坏死核心扩大及纤维帽变薄等AS斑块的不稳定特征。在体外研究中,该团队发现SBK2的缺失促进了巨噬细胞NLRP3炎症小体的激活及细胞因子IL-1β和IL-18的分泌。在机制研究中,该团队发现SBK2与NLRP3存在直接相互作用。体外激酶实验结合Phos-tag及质谱分析发现,SBK2可催化NLRP3蛋白第161位丝氨酸(Ser161)发生磷酸化修饰,进而促进NLRP3通过Tollip介导的选择性自噬途径发生降解,最终抑制NLRP3炎症小体的激活。最后,该团队采用化合物库虚拟筛选的方法,发现了天然化合物RN是SBK2的高效特异性激动剂。随后在细胞和动物水平,验证了RN可剂量依赖性地抑制NLRP3炎症小体激活和L-1β/IL-18分泌,并减少了小鼠的AS斑块负荷。以上研究揭示了蛋白激酶SBK2通过磷酸化修饰调控NLRP3蛋白稳态的新机制,为AS炎症机制研究提供了新视角,为ASCVD抗炎治疗提供了潜在的新靶点。该研究发表于European Heart Journal,山东大学齐鲁医院心血管内科博士生蔡亮宇是该文的第一作者,张澄教授、张猛教授为该文的共同通讯作者。山东大学齐鲁医院为第一和通讯作者单位。
目前,AMI仍是全球人群死亡的主要原因,尽管AMI早期血运重建治疗和指南导向的药物治疗已显著降低了AMI患者的住院期死亡率,但出院患者心力衰竭和心源性猝死的发病率仍居高不下,成为一个重大的临床挑战。在AMI发生过程中,M1型巨噬细胞可过度分泌炎性细胞因子而加剧心肌损伤,调控M1型巨噬细胞的活性可能成为AMI的新型治疗策略。已有研究显示,蛋白质甲基供体SAM与心血管功能异常密切相关。然而,导致蛋白质发生甲基化修饰的蛋白质精氨酸甲基转移酶(PRMTs)在AMI发生和发展中的作用尚不明确。
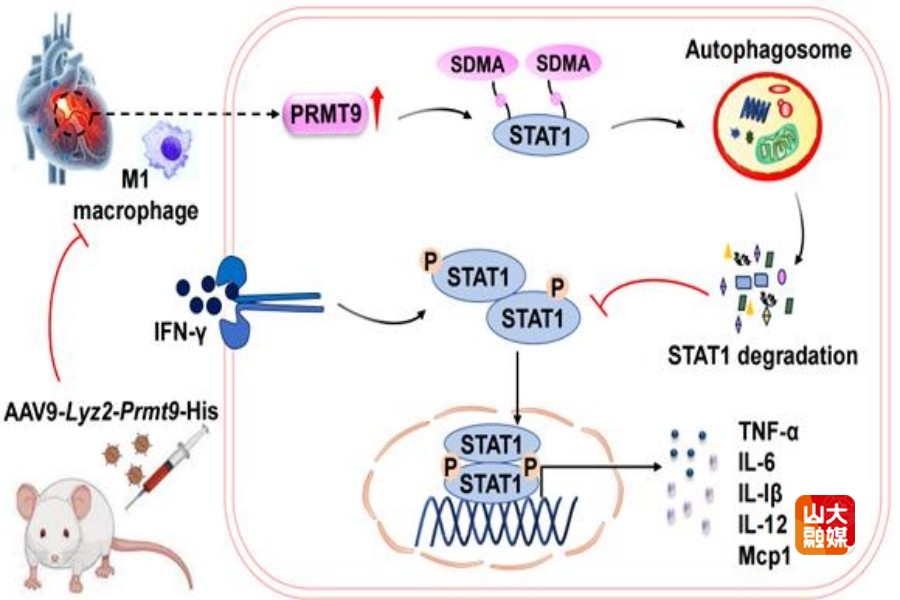
为了回答以上问题,齐鲁医院全国重点实验室张猛教授团队与山东大学基础医学院免疫系高成江教授和刘冰玉教授团队开展了合作研究,该团队通过对单细胞测序和RNA-seq数据集的分析,发现PRMT9在心肌梗死急性期巨噬细胞中的表达特异性升高,该趋势在小鼠心脏巨噬细胞及患者外周血单个核细胞中均得到验证。为了探讨PRMT9在心肌梗死后心脏功能中的作用,研究者在PRMT9基因敲除小鼠中构建了心肌梗死模型,发现巨噬细胞中PRMT9缺失加剧了心肌梗死后的心功能异常,表现为梗死面积扩大、射血分数降低、心肌重构加重及存活率下降。进一步研究显示,PRMT9基因敲除小鼠心脏组织及巨噬细胞中M1型标志基因(Il-6、Tnf-a、Il-1b、Inos)表达及蛋白分泌显著增加。在机制研究中,该团队通过质谱鉴定、免疫共沉淀等技术,证明PRMT9与STAT1可直接结合并催化STAT1蛋白的R588与R736两个精氨酸位点发生甲基化修饰,后者进一步促进STAT1通过自噬-溶酶体途径发生降解,最终抑制巨噬细胞向促炎表型的转换。最后,该团队采用STAT1的药理学抑制剂Fludarabine及AAV9介导的巨噬细胞特异性STAT1敲低两种干预策略,证实抑制STAT1信号可有效逆转PRMT9缺失引起的心肌梗死加重这一不良表型。这一研究结果不仅揭示了在AMI发病过程中PRMT9靶向STAT1的新机制,也为AMI的精准治疗提供了重要的科学依据和潜在干预靶点。该研究发表于Circulation,山东大学基础医学院博士后柏雪梅、博士生袁嘉华和齐鲁医院心血管内科博士生任睿清是该文的共同第一作者,山东大学齐鲁医院张猛教授、山东大学基础医学院免疫系高成江教授和刘冰玉教授为该文的共同通讯作者,山东大学基础医学院和齐鲁医院为共同通讯作者单位。
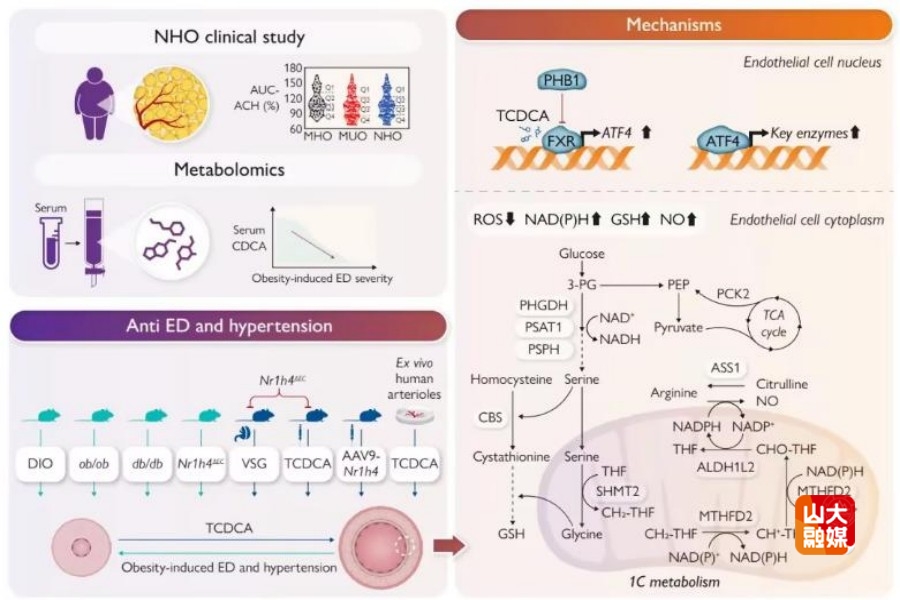
已知血管内皮功能异常是高血压、动脉粥样硬化等心血管疾病的早期和共同发病机制,而肥胖是心血管疾病的危险因素,但不合并心血管疾病的单纯肥胖患者是否存在血管内皮损伤及其分子机制尚不明确。深入探讨单纯肥胖和血管内皮功能异常的关系及其机制,对于防治心血管疾病具有重要的理论和临床价值。
针对上述科学问题,山东大学齐鲁医院全国重点实验室的张文程教授团队在213例不合并高血压的肥胖患者(NHO)中,分离其袖状胃减重手术中切除的大网膜脂肪微动脉,利用wire myograph进行了多维度的血管功能检测。结果发现,代谢健康型(MHO)与代谢不健康型(MUO)肥胖患者的血管内皮功能并无显著性差异且表现出较大的异质性。这一结果挑战了传统的代谢健康肥胖学说,提示MHO可能仅是暂时的过渡阶段,强调对所有肥胖患者的心血管健康均应予以关注。此外,肥胖患者血管内皮功能与传统危险因素并无显著相关,因此研究人员进一步探讨了肥胖患者血管内皮功能的规律特征。通过全定量代谢组学分析,研究人员发现患者血清中的胆汁酸水平,尤其是鹅去氧胆酸(CDCA)与血管内皮功能呈显著正相关,提示CDCA可作为潜在的生物标志物。进一步的离体实验显示,在多种胆汁酸中,CDCA及其牛磺酸结合形式(TCDCA)能显著缓解肥胖相关的内皮功能异常。机制研究证实,胆汁酸核受体FXR在这一过程中起关键作用,而膜受体TGR5则无明显作用,提示血管内皮FXR可作为潜在的治疗靶点。研究人员结合多组学研究,深入揭示了TCDCA-FXR轴的详细机制:TCDCA通过激活血管内皮细胞的丝氨酸代谢及一碳代谢,在转录因子ATF4的介导下,调节肥胖状态下的细胞氧化还原平衡,从而改善了血管内皮功能。此外,研究人员还确定了PHB1作为转录抑制因子参与了FXR对ATF4的调控过程。动物实验结果进一步证实,血管内皮特异性FXR敲除会显著削弱减重手术对肥胖相关血管内皮功能和高血压的治疗效果。同时,TCDCA在小鼠体内展现出良好的降压及改善血管内皮功能的作用,但该效应在内皮细胞敲除FXR的小鼠中消失。该研究首次明确了肥胖患者血管内皮功能的特征,揭示了TCDCA通过FXR受体调控丝氨酸代谢、维持氧化还原平衡,进而缓解内皮功能异常和高血压的分子机制,为肥胖相关心血管疾病的精准防治提供了新的代谢靶点和理论依据。该研究发表于European Heart Journal,山东大学齐鲁医院吕翰林医师、吴智楠博士、万萌博士为本文的共同第一作者,齐鲁医院张文程教授、刘少壮教授和杨建民教授为本文的共同通讯作者。山东大学齐鲁医院为第一和通讯作者单位。